- Home
- > Research
- > Research Highlights
- > Vol. 45 Artificial Intelligence Learns・・・
 Research Highlights
Research Highlights
[Vol. 45]
Artificial Intelligence Learns to Predict Photo-Functional Molecules
- Previous
- Index
- Next
20 Dec, 2018
Artificial intelligence can be used to design new molecules; it is becoming a popular tool because of its potential for discovering molecules in unexplored chemical spaces, its ability to screen a huge number of potential molecules in a short amount of time and its tendency to find unconventional ways of solving problems. However, whether such molecules can be actually synthesized and whether they display the desired functionalities in the real world is an open question.
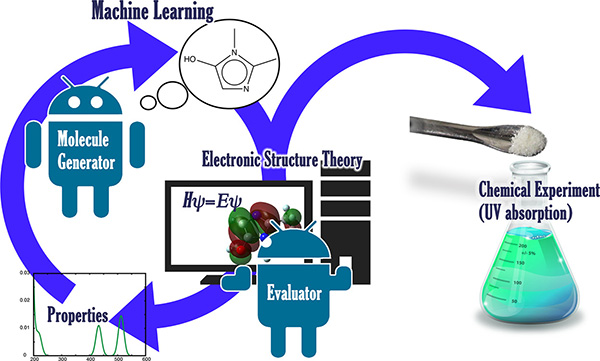
Ryo Tamura, Shinsuke Ishihara at WPI-MANA and colleagues at different institutions in Japan report in ACS Central Science a proof-of-concept study in which they use a platform based on artificial intelligence to discover photo-functional organic molecules (which are relevant in green chemistry and molecular sensing) that can be synthesized and that have specific functional properties. The platform combines a molecule generator powered by artificial intelligence and a calculator based on density functional theory that performs quantum chemical calculations. The generator suggests molecules with different structures, the calculator predicts their properties. As a result, this platform can design new molecules with desired properties.
The system was initially configured to propose molecules with first excited states at five different wavelengths, and it was trained on a database containing 13,000 molecules. After ten days of operation, the algorithm suggested 3,200 different molecules, 86 of which had the first excited level close to one of the desired energies. Six of these molecules were selected for real world synthesis. The selection was done based on two criteria: the existence of at least one known synthetic route to produce them and the transition from the ground state to the first excited state being energetically possible. Five of the six molecules displayed in spectroscopic measurements a first excited state at the desired energy; the other molecule perhaps underwent a decomposition process and did thus not work as predicted.
Normally, to tweak the absorption properties of a molecule chemists alter the structure to modify transitions from the π to the π* orbitals, but, surprisingly, the system mostly suggested molecules in which the relevant transition happens from the non-bonding n orbital to the π* orbital. “This illustrates AI-chemistry’s ability to not only accelerate discovery, but also shed light on hidden paths of possible research,” comment the authors. Because the system relies on property prediction by density functional theory, it inherits the drawbacks of this method, in particular the tendency of underestimating the excitation energy. Thus, there is still work to do, but the potential is high for a transformative tool in chemistry.
Reference
"Hunting for Organic Molecules with Artificial Intelligence: Molecules Optimized for Desired Excitation Energies"
Masato Sumita, Xiufeng Yang, Shinsuke Ishihara, Ryo Tamura, and Koji Tsuda
Journal : ACS Cent. Sci. 4, 1126−1133 (2018).
DOI : 10.1021/acscentsci.8b00213
Masato Sumita, Xiufeng Yang, Shinsuke Ishihara, Ryo Tamura, and Koji Tsuda
Journal : ACS Cent. Sci. 4, 1126−1133 (2018).
DOI : 10.1021/acscentsci.8b00213
Affiliations
International Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), Namiki 1-1, Tsukuba, Ibaraki 305-0044, Japan
Contact information
Research Center for Materials Nanoarchitectonics (MANA)
National Institute for Materials Science
1-1 Namiki, Tsukuba, Ibaraki 305-0044 Japan
Phone: +81-29-860-4710
E-mail: mana-pr[AT]nims.go.jp
1-1 Namiki, Tsukuba, Ibaraki 305-0044 Japan
Phone: +81-29-860-4710
E-mail: mana-pr[AT]nims.go.jp