Large-scale calculation capable of handling material systems containing 100 to 1,000 times more atoms than conventional methods.
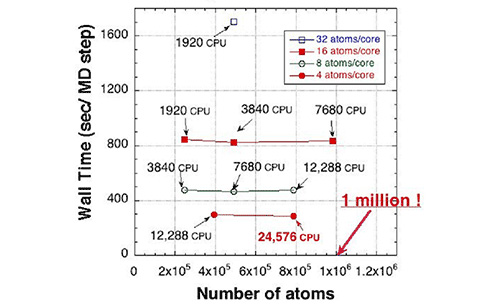
Figure 1: Calculation time of O(N) first-principles calculation program run by the “K computer” (1 CPU = 8 cores). The calculations were performed on silicon systems. The horizontal axis shows the number of atoms, indicating that first-principles calculation of million atom systems is possible. The O(N) method combined with ideal parallel performance of the program (twice the number of CPUs achieving twice the amount of calculation) can calculate systems containing twice the number of atoms in the same time by doubling the number of CPUs.
Calculation time by “K computer”
Elapsed time (sec/step)
Number of atoms
16 atoms/core
8 atoms/core
4 atoms/core
Million atoms
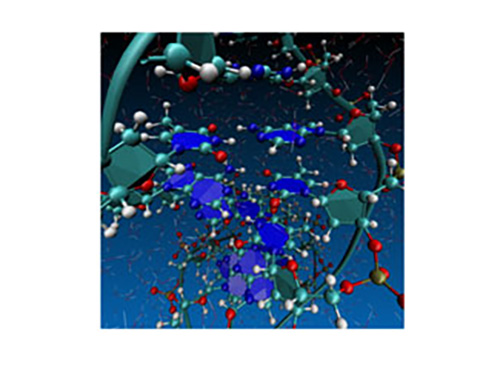
Figure 2: Snapshot structure from first-principles simulation of DNA in water medium using the calculation method developed by the research. The forces between atoms are calculated by first-principles calculation (joint research with RIKEN).
Matter is composed of atoms, and its physical properties are determined by the complex interactions between atoms and electrons. Theoreticians use quantum mechanics to calculate the forces between atoms, and the behaviour of electrons in materials. Specifically, first-principles simulations are based on quantum mechanics, and are a powerful technique widely used to elucidate diverse properties of matter and materials at the atomic scale.
However, the size of the systems modelled with conventional first-principles methods is limited to only a few hundred atoms (in most cases) because the complexity and time required for simulations increases as the cube of the number of atoms being modelled.
Now, a research team led by David Bowler, NIMS MANA and UCL London Centre for Nanotechnology, and Tsuyoshi Miyazaki at NIMS, has successfully developed a highly efficient, large-scale first-principles simulation method for simulating the dynamics of very large systems, containing 100-1,000 times more atoms than conventional methods (up to millions of atoms).
This method provides the means of performing atomic and electronic structure simulations of biological molecules and complex matter, including nanostructured materials, for which conventional methods cannot be used.
The research team has been pursuing the development of a calculation method capable of performing highly efficient large-scale simulations of dynamics. Here, by introducing a new technique where the time required increases linearly with the number of atoms and utilizing supercomputers, namely the “K computer” and FX10 installed at RIKEN and the University of Tokyo, respectively, the team successfully performed first-principles dynamical simulations of systems comprising more than 30,000 atoms, which is 100 times larger than is usual with conventional methods. Their success will pave the way for simulation of very large systems including up to millions of atoms.
"Stable and Efficient Linear Scaling First-Principles Molecular Dynamics for 10,000+ atoms"
Michiaki Arita, David R. Bowler, and
Tsuyoshi Miyazaki
Journal : Journal of Chemical Theory and Computation 10 , 5419 (2014).
DOI :
10.1021/ct500847y
International Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), Namiki 1-1, Tsukuba, Ibaraki 305-0044, Japan
References
"Stable and Efficient Linear Scaling First-Principles Molecular Dynamics for 10,000+ atoms", M. Arita, D. R. Bowler, T. Miyazaki, Journal of Chemical Theory and Computation, 10 , 5419 (2014). DOI: 10.1021/ct500847y
ナノアーキテクトニクス材料研究センター(MANA)
〒305-0044 茨城県つくば市並木1-1
TEL: 029-860-4710
E-mail: mana-pr=ml.nims.go.jp([ = ] → [ @ ] )
To receive our e-mail newsletter “MANA Research Highlights”, please send an e-mail with "MANA Research Highlights request” in the subject line or main text to the following address: mana-pr_at_ml.nims.go.jp
*Please change "_at_ " in the email address to @.
 Research Highlights
Research Highlights