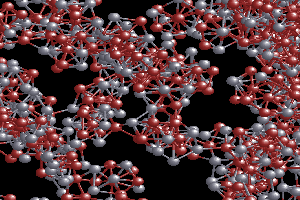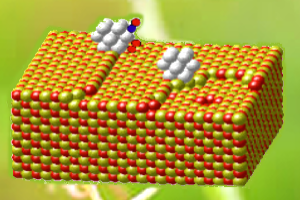
In order to perform reliable simulations on very large systems, such as nano-structured materials, nano-scaled catalysts, and bio-systems, we have been developing a linear-scaling DFT method. With this method, we can employ DFT calculations on systems containing more than tens of thousands of atoms.
Electron transport properties of nano-structures such as atomic wires, molecular wires and defects in thin films have attracted much attention recently. We develop the first-principles calculation method to investigate the transport properties of such nano-structures.