
Surface/Interface Science
Surface Dynamics: Adsorption/Diffusion, H/Si(001)
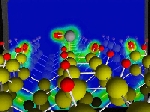 Si atom adsorption on H-terminated Si(001)-(2x1) surface was investigated by using first principles total energy calculations. While it had been considered that H atoms prevent Si adsorption, we found that Si can adsorb on the surface. During the Si adsorption, the Si adatom captures one surface H atom without any energy barrier, forming a SiH structure. It further captures one more surface H atom with a finite energy barrier, forming a SiH2 structure. Adsorbed Si atoms diffuse on the surface with repeating release and capture of H atoms. The diffusion barrier is calculated to be 1.2 eV, which is much larger than that on a bare Si(001) surface.
Si atom adsorption on H-terminated Si(001)-(2x1) surface was investigated by using first principles total energy calculations. While it had been considered that H atoms prevent Si adsorption, we found that Si can adsorb on the surface. During the Si adsorption, the Si adatom captures one surface H atom without any energy barrier, forming a SiH structure. It further captures one more surface H atom with a finite energy barrier, forming a SiH2 structure. Adsorbed Si atoms diffuse on the surface with repeating release and capture of H atoms. The diffusion barrier is calculated to be 1.2 eV, which is much larger than that on a bare Si(001) surface.
Surface Dynamics: O2/Si(001)
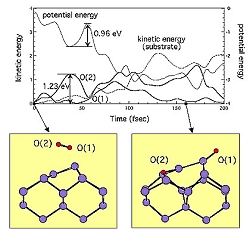 Fig. 1 Kinetic energy analysis of "O(1)" oxygen atom and "O(2)" oxygen atom. The substrate kinetic energy and the potential energy are also shown. The initial oxidation processes on the Si(001) surface are studied by molecular dynamics (MD) simulations based on first-principles (FP) / molecular mechanical (MM) hybrid method [1]. Link molecule method (LMM) is used to couple the FP and the TB region [2]. The backbond oxidation process in the first layer is seen for most of the cases and its mechanism is clarified by the kinetic energy analysis (Fig. 1). The steering motion of the oxygen molecule before dissociation is found in several cases. The backbond oxidation process in the second layer is seen for the cases in which the oxygen molecule is initially located at the site between two neighboring dimer rows.
Fig. 1 Kinetic energy analysis of "O(1)" oxygen atom and "O(2)" oxygen atom. The substrate kinetic energy and the potential energy are also shown. The initial oxidation processes on the Si(001) surface are studied by molecular dynamics (MD) simulations based on first-principles (FP) / molecular mechanical (MM) hybrid method [1]. Link molecule method (LMM) is used to couple the FP and the TB region [2]. The backbond oxidation process in the first layer is seen for most of the cases and its mechanism is clarified by the kinetic energy analysis (Fig. 1). The steering motion of the oxygen molecule before dissociation is found in several cases. The backbond oxidation process in the second layer is seen for the cases in which the oxygen molecule is initially located at the site between two neighboring dimer rows.
References
[1] N. Takahashi, Y. Nakamura, J. Nara, Y. Tateyama, T. Uda and T. Ohno: Surf.Sci.602, pp.768-777 (2008).
[2] Y.Nakamura, N.Takahashi, M.Okamoto, T.Uda, T.Ohno: J.Comp.Phys.225, pp.1985-1993 (2007).
Molecular Interfaces: SAM on Au
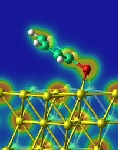 Adsorption of benzenethiol molecules on Au(111) surface was investigated by using first principles total energy calculations. A single thiolate molecule is adsorbed at the bridge site slightly shifted toward the fcc-hollow site, and is tilted by 61° from the surface normal. As for the self-assembled monolayer (SAM) structures, the (2√3x√3)R30° herringbone structure is stabilized against the (√3x√3)R30° structure by large steric relaxation. In the most stable (2√3x√3)R30° SAM structure, the molecule is adsorbed at the bridge site with the tilting angle of 21°, which is much smaller compared with the single molecule adsorption. The van der Waals interaction plays an important role in forming the SAM structure. The adsorption of benzenethiolates induces the repulsive interaction between surface Au atoms, which facilitates the formation of surface Au vacancy.
Adsorption of benzenethiol molecules on Au(111) surface was investigated by using first principles total energy calculations. A single thiolate molecule is adsorbed at the bridge site slightly shifted toward the fcc-hollow site, and is tilted by 61° from the surface normal. As for the self-assembled monolayer (SAM) structures, the (2√3x√3)R30° herringbone structure is stabilized against the (√3x√3)R30° structure by large steric relaxation. In the most stable (2√3x√3)R30° SAM structure, the molecule is adsorbed at the bridge site with the tilting angle of 21°, which is much smaller compared with the single molecule adsorption. The van der Waals interaction plays an important role in forming the SAM structure. The adsorption of benzenethiolates induces the repulsive interaction between surface Au atoms, which facilitates the formation of surface Au vacancy.
AFM: Tip Apex structures QM/MM
Multi-regional hybrid simulations with a concurrent use of fist-principles(FP), tight binding(TB), and molecular mechanical(MM) calculations are performed to study the atomic structure of a clean silicon AFM tip [1]. The tip is modeled by 4750 Si atoms and partitioned into FP(92), TB(833), and MM(3825) regions. We find the formation of a distinct atomic protrusion at the tip apex as shown in the movie (300K, 1.6psec). This study gives the reason why the atomic protrusion exits at the apex despite the fact that the atomic geometry of the very end of the tip is practically uncontrollable in the tip preparation.
References
[1] Y.Nakamura, N.Takahashi, T.Uda, T.Ohno: Phys.Rev.Lett.97, 086103 (2006)
 Research Index
Research Index